 |
Author: Dr. phys. Alexei Kuzmin Institute of Solid State Physics University of Latvia Kengaraga street 8, LV-1063 Riga, Latvia Internet: https://www.dragon.lv/edaca E-mail: a.kuzmin@cfi.lu.lv |
| Preface |
X-ray absorption spectroscopy (XAS) at synchrotron radiation sources is a structural tool providing information on the local atomic and electronic structure around an atom of a particular type. Today XAS is successfully applied to a study of crystalline, nanocrystalline and disordered solids, liquids and gases in a wide range of external conditions defined by temperature, pressure, etc. The size of the region, probed by XAS, depends on the degree of thermal and static disorder present in a material and is limited by the mean-free path of the excited photoelectron. Typically the information reach region extended up to 3-10 A around the absorbing atom. An advantage of the XAS method is its sensitivity to many-atom distribution functions, giving rise to multiple-scattering (MS) contributions, and to correlation effects in atom dynamics. Note that accurate account of both effects is still challenging. The time-scale (about 10-15-10-16 s) of the X-ray absorption process is much shorter than the characteristic time (about 10-13-10-14 s) of thermal vibrations. Therefore, the atoms may be considered as frozen at their instantaneous positions during a single photoabsorption process, and the total experimentally measured X-ray absorption spectrum corresponds to the configurational average of all atomic positions over the time of the experiment. This situation can be straightforwardly modelled combining the molecular dynamics (MD) simulation with the extended X-ray absorption fine structure (EXAFS) calculations, known as the MD-EXAFS approach. Finally, the agreement between the experimental and configuration-averaged EXAFS spectra can be used to validate the accuracy of the interatomic potential (force-field) models employed in the MD simulations. |
||||||||||||||||||||||||||
| MD-EXAFS method |
The general scheme of the MD-EXAFS method is shown in figure below. 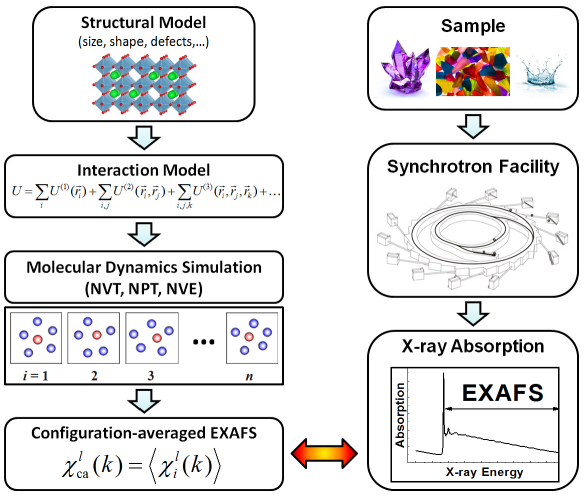
First, the structural model of the system should be constructed, taking into account its periodicity, the presence of defects as well as size and shape in the case of nanocrystals and clusters. Example of the experimental (black open circles) and configuration-averaged (blue solid lines) EXAFS spectra for the Ti K-edge in SrTiO3 and the Cu K-edge in fcc Cu are shown below: 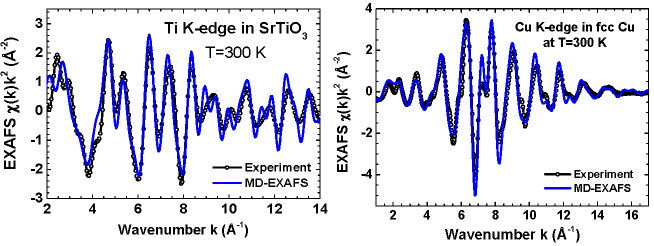 |
||||||||||||||||||||||||||
| Configuration averaged EXAFS calculations |
The EDACA code is composed of three programs edamd.exe, edaca.exe and stdev.exe.
edamd.exe Fe_bcc_300K_MD.xyz 0 0 8.0 Fe 0 As an alternative, the three programs can be run also separately without employing the runedaca.bat file. The edamd.exe code requires six or more parameters to be specified: edamd.exe filename_xyz SkipFirst Skip Rmax atom1 absorber_number atom2 atom3 ...
The order of elements in the command line is important and should correspond to that in the pot.dat file (see below), the first atom is always the absorber. The edaca.exe code uses the results produced by the edamd.exe code plus a number of additional files, which should be located in the same directory.
A set of files required by the edaca.exe includes: The edaca.exe code calculates EXAFS spectrum for each XYZ file specified in the conf.dat file. These spectra are saved under the names xt.001, xt.002, …. The main result is saved under the name xt_tot.txt and contains the configuration-averaged EXAFS spectrum. The configuration-averaged X-ray absorption coefficient is also saved under the name mu_tot.txt. |
||||||||||||||||||||||||||
| Downloads |
Download EDACA User's Manual in PDF format.
Download EDACA package for Windows.
Download EDACA package for Linux. |
||||||||||||||||||||||||||
| Presentations |
A. Kuzmin, The use of Molecular Dynamics simulations for the interpretation of EXAFS spectra. Lecture at Brookhaven National Laboratory, November 1-3, 2023. A. Kuzmin, EDACA software demonstration for Molecular Dynamics simulations of EXAFS spectra. Lecture at Brookhaven National Laboratory, November 1-3, 2023. |
||||||||||||||||||||||||||
| References |
Please cite these works in your publications based on the results of the EDACA simulations: A. Kuzmin, R.A. Evarestov, Quantum mechanics-molecular dynamics approach to the interpretation of X-ray absorption spectra, J. Phys.: Condens. Matter 21 (2009) 055401 (6 pp). DOI: 10.1088/0953-8984/21/5/055401 A. Kuzmin, A. Anspoks, A. Kalinko, J. Timoshenko, The use of x-ray absorption spectra for validation of classical force-field models, Z. Phys. Chem. 230 (2016) 537-549. DOI: 10.1515/zpch-2015-0664 |