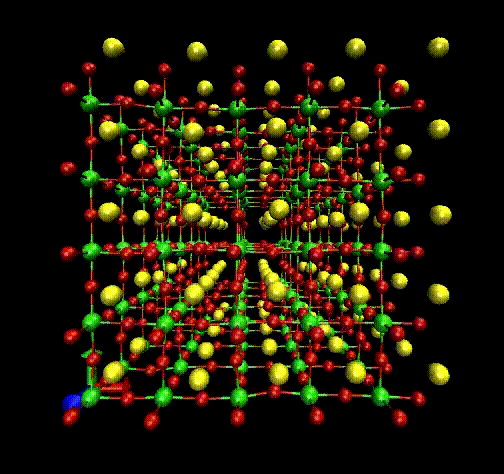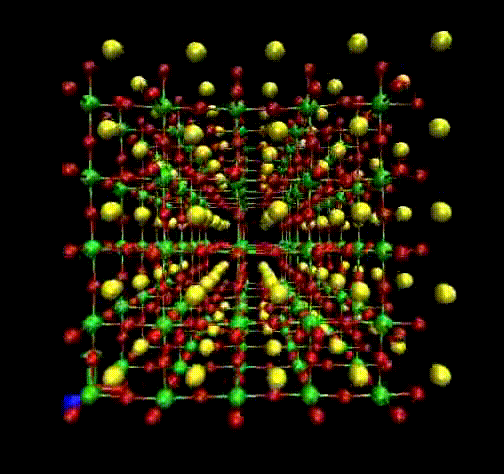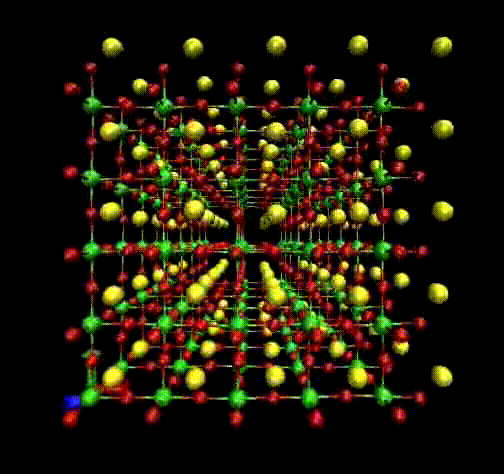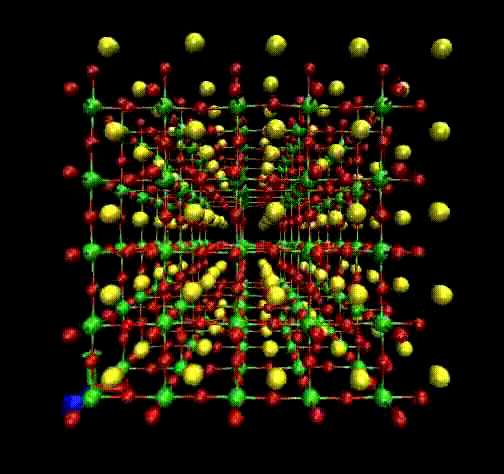
Molecular Dynamics (MD) is a theoretical simulation method for studying classical statistical
mechanics of well-defined systems through numerical solutions of Newton's equations of motion.
In our laboratory, we use it to perform interpretation of Raman and EXAFS spectra.
In the EXAFS spectra case, the use of MD allows us to overcome problems related to
accurate accounting for disorder effects, in particular, beyond the first coordination
shell - in the region, where the multiple-scattering (MS) effects, depending on many-atom
distributions, contribute.
Other examples:
A. Anspoks, A. Kalinko, R. Kalendarev, A. Kuzmin,
Atomic structure relaxation in nanocrystalline NiO studied by EXAFS spectroscopy: Role of nickel vacancies,
Phys. Rev. B 86 (2012) 174114:1-11.
J. Timoshenko, A. Kuzmin, J. Purans,
Molecular dynamics simulations of EXAFS in germanium,
Centr. Eur. J. Phys. 9 (2011) 710-715.
A. Kuzmin, V. Efimov, E. Efimova, V. Sikolenko, S. Pascarelli, I. O. Troyanchuk,
Interpretation of the Co K-edge EXAFS in LaCoO3 using molecular dynamics simulations,
Solid State Ionics 188 (2011) 21-24.
A. Kalinko, R.A. Evarestov, A. Kuzmin, J. Purans,
Interpretation of EXAFS in ReO3 using molecular dynamics simulations,
J. Phys.: Conf. Ser. 190 (2009) 012080 (4pp).
USEFUL LINKS
Wikipedia: Molecular dynamics.
GULP - the General Utility Lattice Program.
LAMMPS - a Large-scale Atomic/Molecular Massively Parallel Simulator.
HOOMD-blue - a general-purpose particle simulation toolkit.
DL_POLY - a general purpose serial and parallel molecular dynamics simulation package.
IMD - a software package for classical molecular dynamics simulations.
XMD - Molecular Dynamics for metals and ceramics.
OpenMD - an open source molecular dynamics engine.
PIMD - an open-source software for parallel molecular simulations.
XenoView - a Windows based software for molecular dynamics simulations..
CP2K - a program to perform atomistic and molecular simulations of solid state, liquid, molecular, and biological systems.
CPMD - a parallelized plane wave / pseudopotential implementation of Density Functional Theory, designed for ab-initio molecular dynamics.
OpenKIM - the knowledgebase of interatomic models.
Database of Published Interatomic Potential Parameters.
Interatomic Potentials Repository Project at NIST.
JARVIS Force-fields at NIST.
Atomicrex - a tool for the construction of interaction models.
atsim.potentials - a tool for potential model tabulation.
Potfit - a free implementation of the force-matching algorithm to generate effective potentials from ab-initio reference data.
GARFfield - a multi-platform, multi-objective parallel hybrid genetic algorithm (GA) / conjugate-gradient (CG) based force field optimization framework.
PACKMOL creates an initial point for molecular dynamics simulations by packing molecules in defined regions of space.
VESTA - a 3D visualization program for structural models and 3D grid data such as electron/nuclear densities.
VMD - a molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting.
Ovito - scientific visualization and analysis software for atomistic simulation data.
MOLEKEL - an open-source multi-platform molecular visualization program.
GDIS - a GTK based program for the display and manipulation of isolated molecules and periodic systems.
Travis - trajectory analyzer and visualizer.
Atomsk - a command-line program that aims at creating, manipulating, and converting atomic systems, for the purposes of ab initio calculations, classical potential simulations, or visualization.
POV-Ray - the Persistence of Vision Raytracer, a tool for producing high-quality computer graphics.
Engineering Virtual Organization for Cyber Design.
Materials Square.
Accelrys.
LAMMPS tutorials.
|