Applications
See also our review papers:
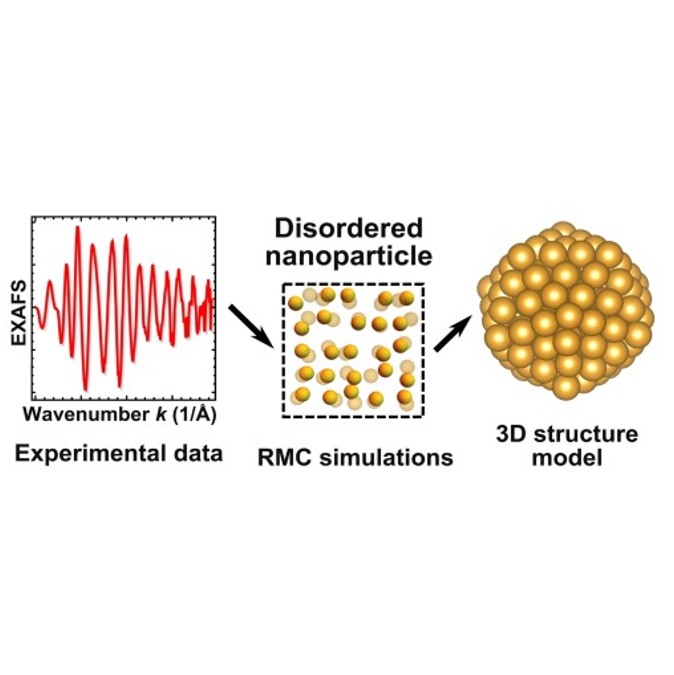
Solving the structure and dynamics of metal nanoparticles by combining X-ray absorption fine structure spectroscopy and atomistic structure simulations
J. Timoshenko, Z. Duan, G. Henkelman, R.M. Crooks, A.I. Frenkel, Annu. Rev. Anal. Chem. 12 (2019) 501-522.
Treatment of disorder effects in X-ray absorption spectra beyond the conventional approach
A. Kuzmin, J. Timoshenko, A. Kalinko, I. Jonane, A. Anspoks, Rad. Phys. Chem. 175 (2020) 108112
In situ/operando electrocatalyst characterization by X-ray absorption spectroscopy
J. Timoshenko and B. Roldan Cuenya, Chem. Rev. 121 (2021) 2021 882
***
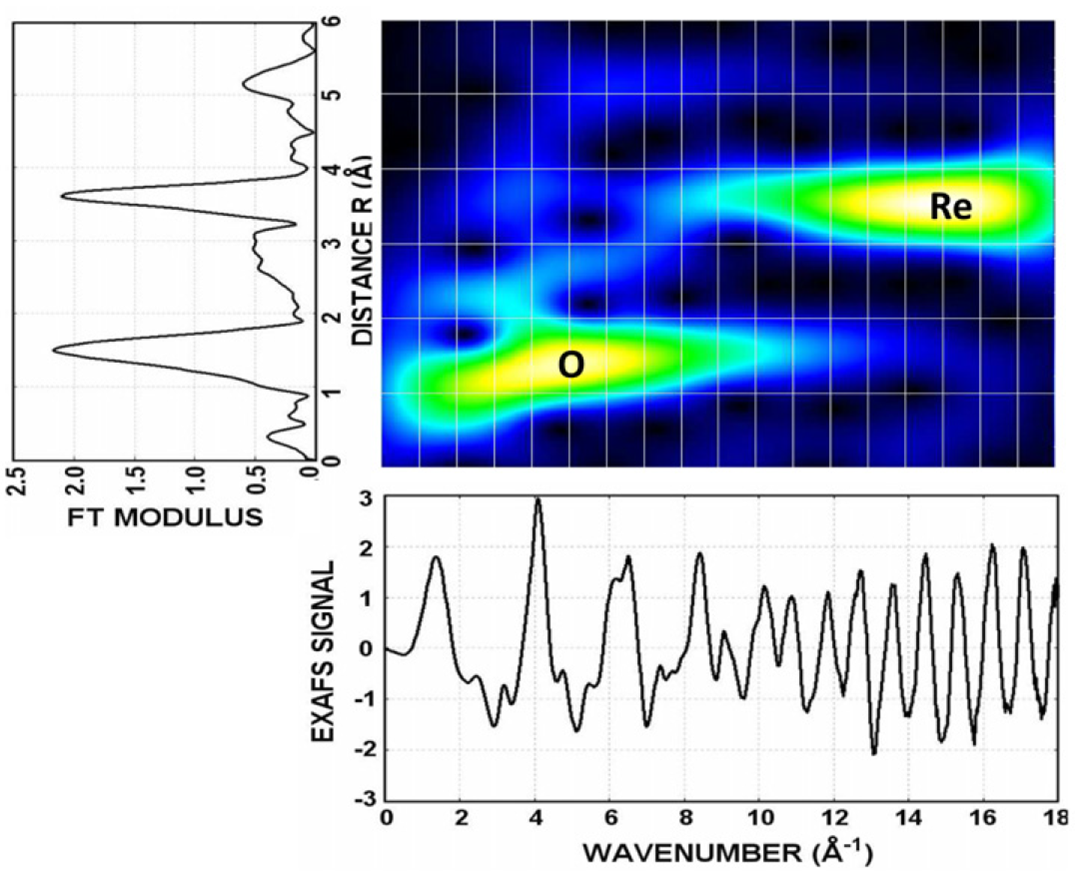
Wavelet data analysis of EXAFS spectra
J. Timoshenko and A. Kuzmin, Comp. Phys. Commun. 180 (2009) 920-925.
The application of wavelet transform to the analysis of the EXAFS signals of perovskite-type compounds is demonstrated. We found that the wavelet transform
is advantageous method compared with conventional Fourier transform both for visualization and interpretation of the EXAFS
spectra, especially, for compounds composed of heavy and light elements.
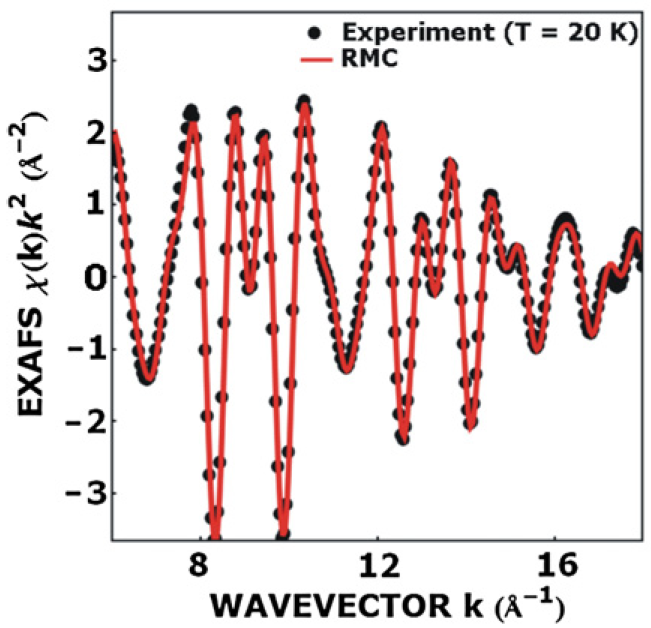
Reverse Monte Carlo modelling of thermal disorder in crystalline materials from EXAFS spectra
J. Timoshenko, A. Kuzmin, J. Purans, Comp. Phys. Commun. 183 (2012) 1237-1245.
The improved Reverse Monte Carlo (RMC) scheme for the analysis of the EXAFS spectra from crystalline materials is presented. The use of the method is demonstrated on the example of the
EXAFS spectra analysis for the model system and experimental data for crystalline germanium and rhenium trioxide.
An efficient implementation of the reverse Monte Carlo method for EXAFS analysis in crystalline materials
J. Timoshenko, A. Kuzmin, J. Purans, J. Phys.: Conf. Ser. 430 (2013) 012012:1-4.
The ability of RMC method to interpret EXAFS spectra of crystalline materials even in case, when the multiple-scattering
effects are very pronounced, is demonstrated. The analysis of the Re L3-edge EXAFS data from the second
and third coordination shells of rhenium in ReO3 has been carried out for the first time. We affirm the strong
correlation between displacements of oxygen and nearest rhenium atoms but also the strong correlation between the motion of two
nearest rhenium atoms is revealed.
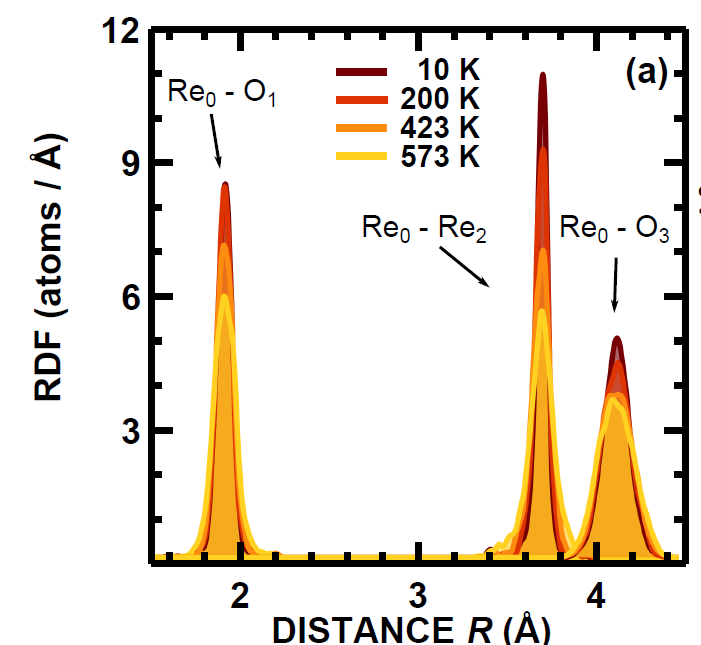
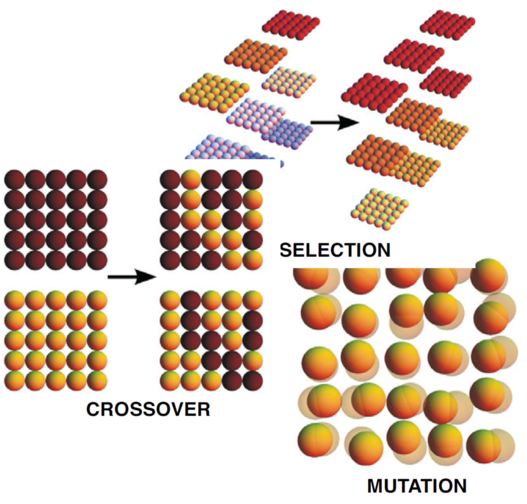
EXAFS study of hydrogen intercalation into ReO3 using the evolutionary algorithm
J. Timoshenko, A. Kuzmin, J. Purans, J. Phys.: Condens. Matter 26 (2014) 055401 (15pp).
The superiority of evolutionary algorithm (EA) over conventional RMC scheme is demonstrated. The novel simulation approach, based on the use of EA, allowed us to follow structure development at different steps of hydrogen intercalation into ReO3 from in situ EXAFS measurements at the Re L3edge. We found that atomic motion is strongly correlated in the
first and second coordination shells of rhenium in cubic ReO3,
whereas it becomes significantly less correlated in HxReO3.
The changes in the correlation of atomic motion are attributed
to an electronic origin, since they have a short relaxation time.
This effect has been related to additional electrons, localized
largely at rhenium atoms and entering into the crystal lattice
together with hydrogen ions to maintain charge neutrality.
Besides, the insertion of hydrogen atoms leads to a
distortion and tilting of the rhenium–oxygen octahedra. Our
analysis revealed that these processes have significantly longer
characteristic times.
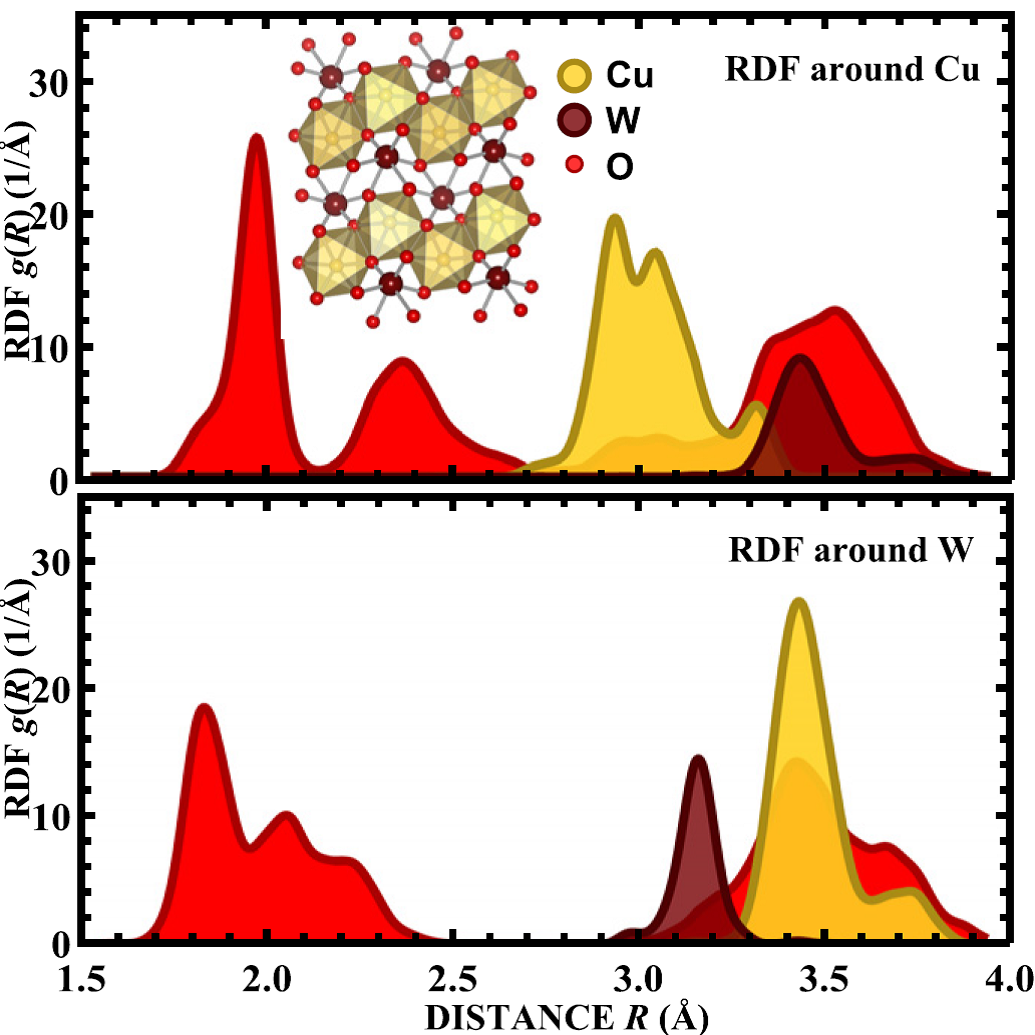
Analysis of EXAFS data from copper tungstate by reverse Monte Carlo method
J. Timoshenko, A. Anspoks, A. Kalinko, A. Kuzmin, Phys. Scripta 89 (2014) 044006 (6pp).
The analysis of temperature-dependent Cu K and W L3-edges EXAFS spectra for CuWO4 has been performed. RMC simulations using EXAFS data for both
edges simultaneously allowed us to reconstruct the local structure around both the Cu atoms and W atoms. The analysis of
the first coordination shell around Cu atoms revealed that the distribution of the nearest four oxygen atoms is nearly
temperature independent due to strong Cu–O bonds, while the distribution of remaining two oxygen atoms broadens upon
increasing temperature.
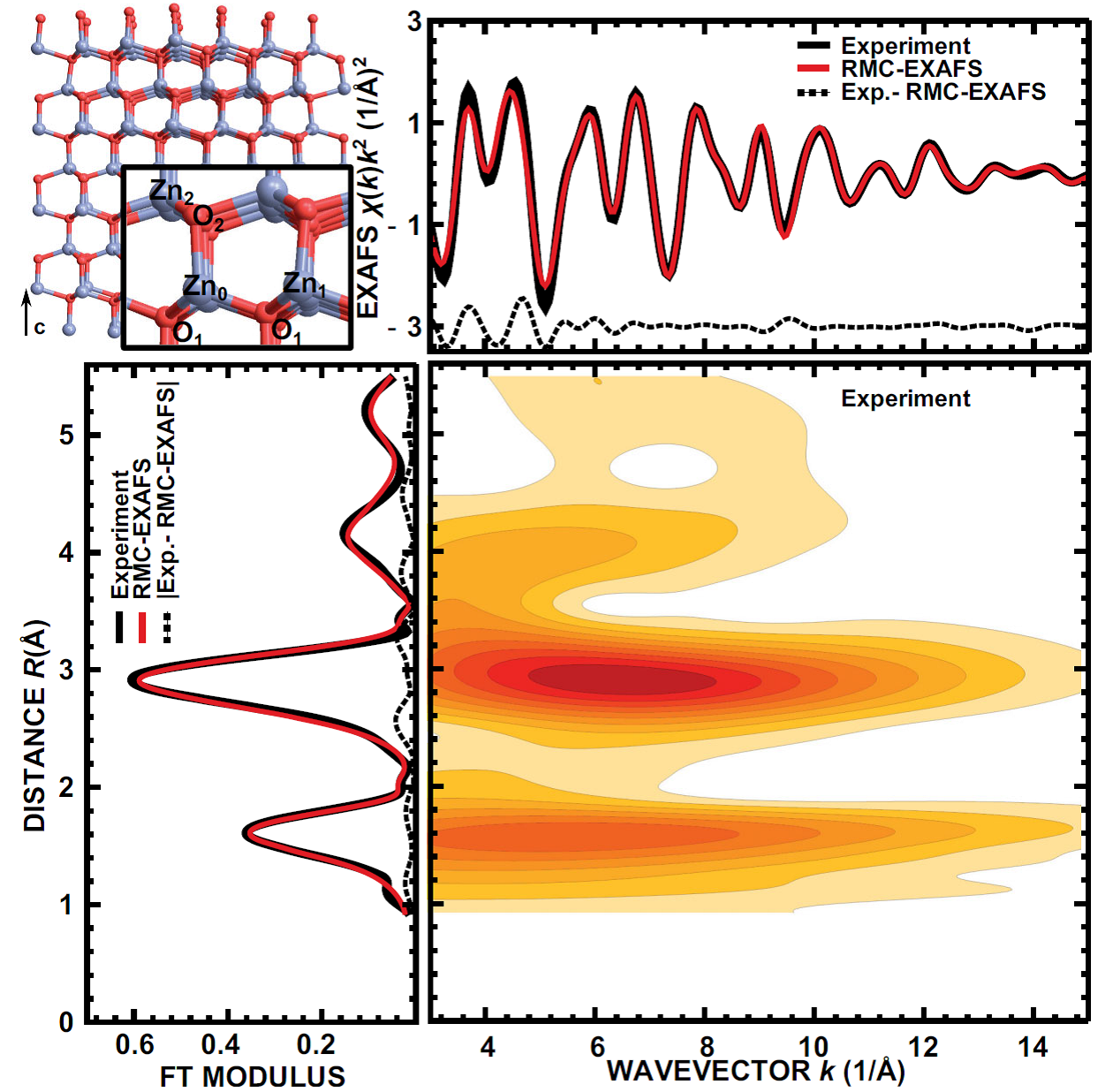
Local structure and dynamics of wurtzite-type ZnO from simulation-based EXAFS analysis
J. Timoshenko, A. Anspoks, A. Kalinko, A. Kuzmin, Phys. Status Solidi (c) 11 (2014) 1472-1475.
Room temperature Zn K-edge EXAFS spectrum was analysed using two advanced theoretical
approaches based on classical molecular dynamics
(MD) and reverse Monte Carlo/evolutionary algorithm
(RMC/EA) methods. The RMC/EA-EXAFS
method allowed us to discriminate contributions of nonequivalent
Zn and O atoms in the first and second coordination
shells around absorbing Zn atom. The obtained
results revealed essential differences between Zn–O and
Zn–Zn bonds, aligned along the c-axis direction and in the
orthogonal direction of ZnO hexagonal crystal lattice.
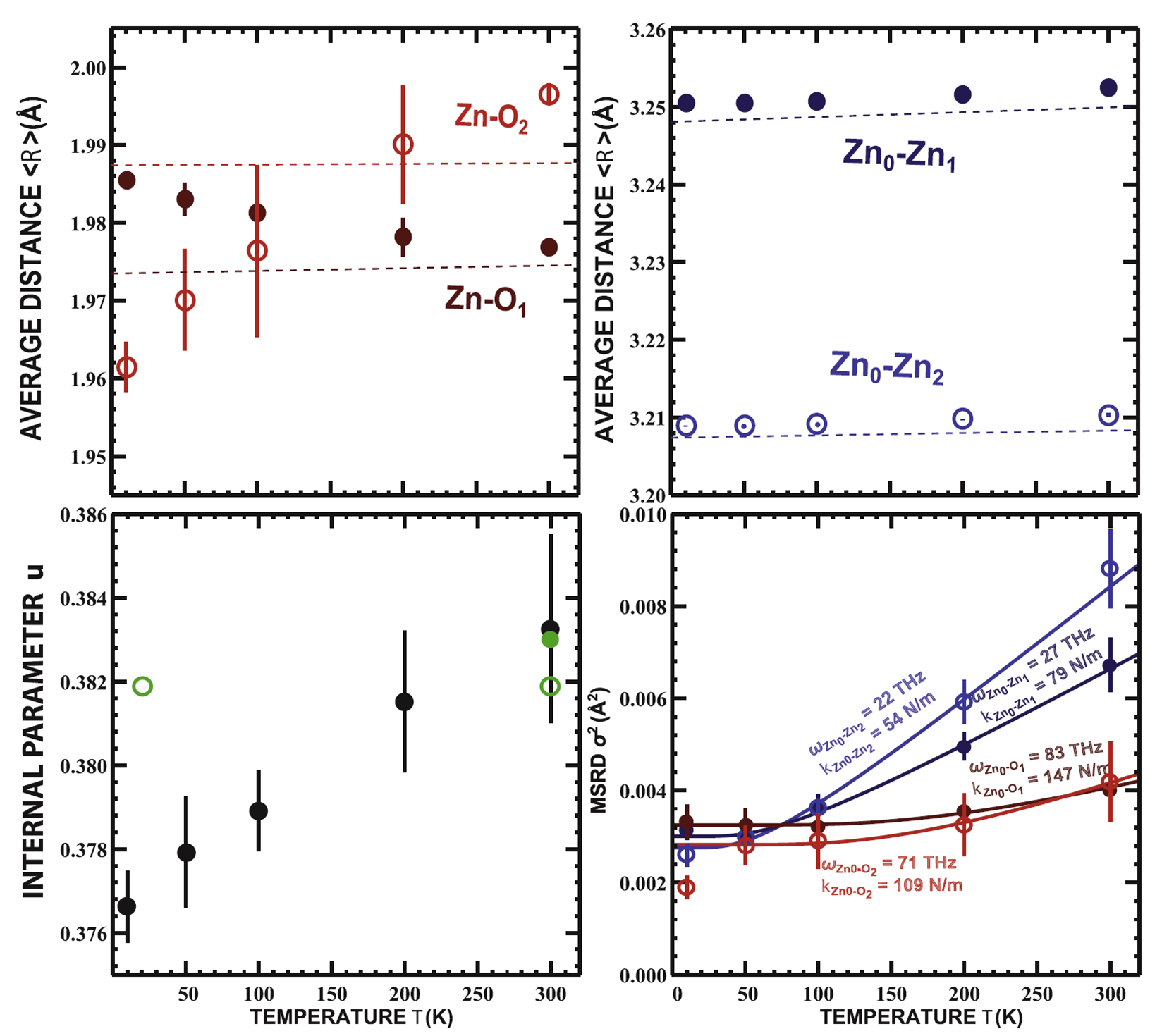
Temperature dependence of the local structure and lattice dynamics of wurtzite-type ZnO
J. Timoshenko, A. Anspoks, A. Kalinko, A. Kuzmin, Acta Mater. 79 (2014) 194-202.
The local atomic structure and lattice dynamics in polycrystalline
wurtzite-type ZnO were studied by the Zn K-edge
X-ray absorption spectroscopy in the temperature
range from 10 to 300 K. Temperature-dependent EXAFS
spectra were analyzed using MD-EXAFS and RMC/EA-EXAFS methods. The RMC/EA-EXAFS method gives a more accurate
description of the thermal disorder in ZnO, since it is not
limited by any simplified model. Our results suggest that both Zn and O atoms, located
within one ab-plane, interact more strongly than the atoms
at approximately the same distance, but located in neighboring
layers along the c-axis. The observed changes in
the average Zn-O distances also imply that with increasing temperature the structure of
ZnO becomes more anisotropic, and the corresponding
internal parameter u of the oxygen Wyckoff position (2b)
increases.
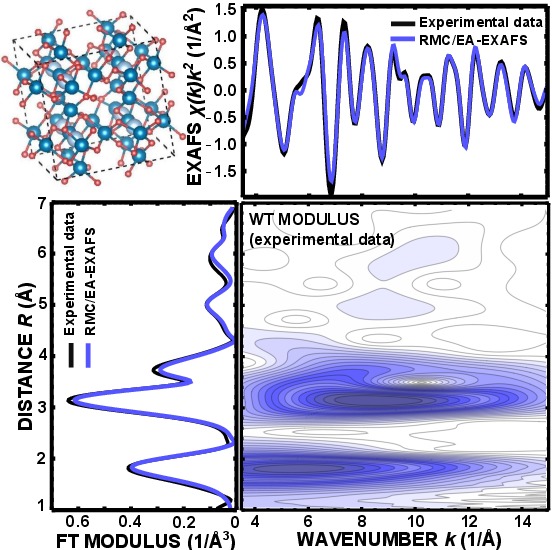
Local structure of nanosized tungstates revealed by evolutionary algorithm
J. Timoshenko, A. Anspoks, A. Kalinko, A. Kuzmin, Phys. Status Solidi A 212 (2015) 265-273.
In order to fully exploit the structural information contained in X-ray absorption
spectra, in this study we employ the evolutionary
algorithm (EA) approach for the interpretation of the Co and Cu K-edges
as well as the W L3-edge extended X-ray absorption fine
structure of nanosized CoWO4 and CuWO4. The
combined RMC/EA-EXAFS approach and simultaneous analysis of
the W L3 and Co(Cu) K-edge EXAFS spectra allowed us for
the first time to obtain a 3D structure model of the tungstate
nanoparticles and to explore in details the effect of size,
temperature and transition metal type.
EXAFS study of the local structure of crystalline and nanocrystalline Y2O3 using evolutionary algorithm method
I. Jonane, J. Timoshenko, A. Kuzmin, IOP Conf. Ser.: Mater. Sci. Eng. 77 (2015) 012030:1-5.
Temperature-dependent local structure and lattice dynamics of yttria (Y2O3) is
studied by X-ray absorption spectroscopy. Combination of the reverse Monte
Carlo and evolutionary algorithm techniques is applied for the analysis of extended X-ray
absorption fine structure at the Y K-edge. This approach allows us to reconstruct 3D atomic
structure models of crystalline and nanocrystalline Y2O3.
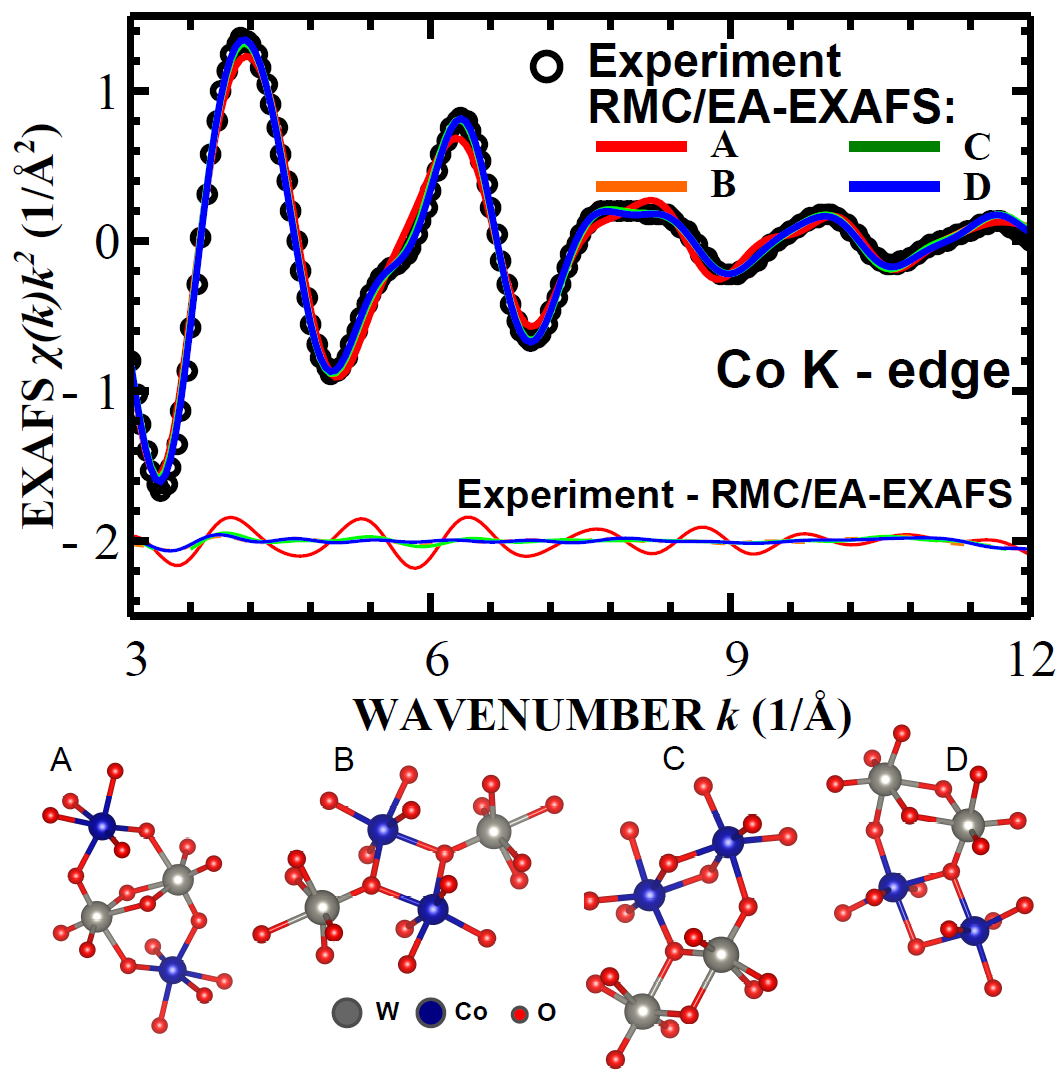
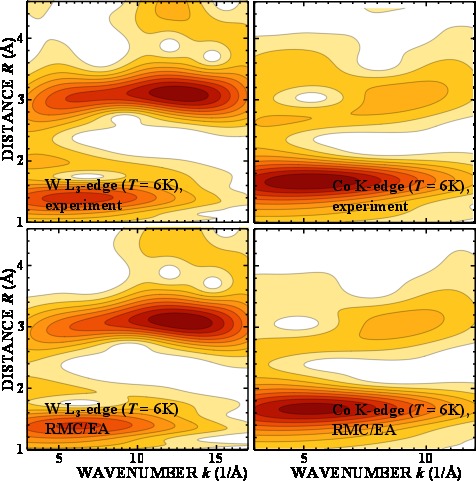
Local structure of cobalt tungstate revealed by EXAFS spectroscopy and reverse Monte Carlo/evolutionary algorithm simulations
J. Timoshenko, A. Anspoks, A. Kalinko, A. Kuzmin, Z. Phys. Chem. (2015), doi: 10.1515/zpch-2015-0646
Reverse Monte Carlo simulations, coupled with the evolutionary algorithm and
wavelet transform are applied to the analysis of the W L3-edge and Co K-edge EXAFS spectra of crystalline CoWO4, which exists in
antiferromagnetic state below 55 K. Temperature dependence of the local environment up to 0.43 nm around
both metal ions is reconstructed in the range from 10 K to 300 K, and the rigidity of the tungstate
structure due to zigzag chains of WO6 and CoO6 octahedra is analyzed.
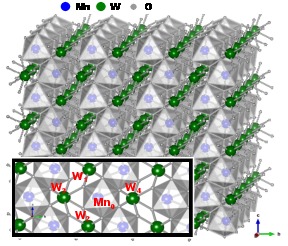
Local structure of multiferroic MnWO4 and Mn0.7Co0.3WO4 revealed by the evolutionary algorithm
J. Timoshenko, A. Anspoks, A. Kalinko, I. Jonane, A. Kuzmin, Ferroelectrics 483 (2015) 68-74
We employ wavelet analysis and RMC/EA simulations to interpret EXAFS spectra in solid solutions
Mn1-xCoxWO4. It is shown that the high-frequency part of the W L3 and Mn(Co) K-edge EXAFS spectra is dominated by contributions from
the Mn(Co)-W atom pairs, and is sensitive to temperature and composition. Essential differences in the dynamics of the Mn-W atom pairs, located along the crystallographic
b- and c-axes, are obtained: the interactions along the c-axis, corresponding to the direction
of zigzag chains of edge-shared MnO6 octahedra, appear to be stiffer.
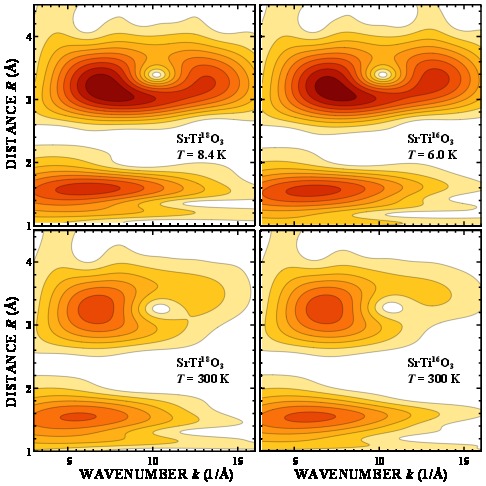
Local structure studies of Ti for SrTi16O3 and SrTi18O3 by advanced x-ray absorption spectroscopy data analysis
A. Anspoks, J. Timoshenko, D. Bocharov, J. Purans, F. Rocca, A. Sarakovskis, V. Trepakov, A. Dejneka, M. Itoh, Ferroelectrics 485 (2015) 42-53
In this work the local atomic structure around Ti in SrTi16O3 and SrTi18O3 is studied in the temperature range 8 – 300 K with X-ray absorption spectroscopy methods, namely
extended x-ray absorption fine structure (EXAFS), advanced reverse Monte Carlo (RMC) and evolutionary algorithm (EA) techniques for EXAFS data analysis (RMC/EA-EXAFS). Obtained results show non-Gaussian character of the RDF for the first three coordination
shells of Ti, especially the first shell in the whole temperature range studied (8 – 300 K).
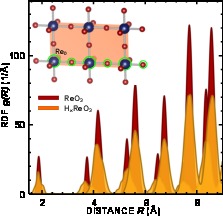
Disappearance of correlations in the atom motion upon hydrogen intercalation into ReO3 lattice
J. Timoshenko, A. Kuzmin, J. Purans, J. Phys.: Conf. Ser. 712 (2016) 12003-12006
In this work the ability to accurately analyze contributions to EXAFS spectra from atoms located at distances up to 0.8 nm from absorbing atom is demonstrated for the first time in ReO3 and HxReO3.
For this purpose we employed a complex approach, based on the use of reverse Monte Carlo method, evolutionary algorithm and wavelet transform. This approach allowed us to reveal the
presence of very strong and relatively long-range correlations in the motion of Re and O atoms within linear -Re-O-Re- chains in pure ReO3. It was also demonstrated that these correlations
are destroyed by the intercalation of hydrogen ions into crystalline ReO3 lattice.
Probing structural relaxation in nanosized catalysts by combining EXAFS and reverse Monte Carlo methods
J. Timoshenko, A.I. Frenkel, Catal.Today. 280 (2017) 274-282
In this work we use RMC/EA method to deal with the asymmetric bond-length distributions in the small metal nanoparticles (1-2 nm). Strain-induced structure relaxation, large structural disorder and complex non-Gaussian shape of the
interatomic distances distribution are characteristic even for the simplest monometallic nanocatalysts,
and may have significant impact on many properties, including catalytic ones. The EXAFS studies of disorder effects in
small nanoparticles are,however, challenging due to the assumption of quasi-Gaussian
bond length distributions that is made in the conventional analysis. Reverse Monte Carlo and evolutionary algorithm methods do not have such limitations. In addition, we use here classical molecular dynamics
simulations to validate the obtained results.
Determination of bimetallic architectures in nanometer-scale catalysts by combining molecular dynamics simulations with x-ray absorption spectroscopy
J. Timoshenko, K.R. Keller, A.I. Frenkel, J. Chem. Phys.146 (2017) 114201.
Here we present an approach for the determination of an atomic structure of small bimetallic nanoparticles
by combining extended X-ray absorption fine structure spectroscopy and classical molecular
dynamics simulations based on the Sutton-Chen potential. The proposed approach is illustrated in
the example of PdAu nanoparticles with ca 100 atoms and narrow size and compositional distributions.
Using a direct modeling approach and no adjustable parameters, we were able to reproduce
the size and shape of nanoparticles as well as the intra-particle distributions of atoms and metal mixing
ratios and to explore the influence of these parameters on the local structure and dynamics in
nanoparticles. RMC/EA simulations are then used to fine-tune the structure models, obtained in MD simulations.

Supervised machine-learning-based determination of three-dimensional structure of metallic nanoparticles
J. Timoshenko, D. Lu, Y. Lin, A.I. Frenkel, J. Phys. Chem. Lett. 8 (2017) 5091−5098.
Here we present another novel method for determination of 3D structure in metallic catalysts, based on analysis of XANES data and supervised machine learning (SML). SML is used to unravel
the hidden relationship between the XANES features and catalyst geometry. To train our
SML method, we rely on ab initio XANES simulations. Our approach allows one to solve
the structure of a metal catalyst from its experimental XANES, as demonstrated here by
reconstructing the average size, shape, and morphology of platinum
nanoparticles. RMC/EA simulations are used here to fine-tune the structure models, obtained in SML/XANES analysis.
Probing atomic distributions in mono- and bimetallic nanoparticles by supervised machine learning
J. Timoshenko, C. J. Wrasman, M. Luneau, T. Shirman, M. Cargnello, S. R. Bare, J. Aizenberg, C. M. Friend, A. I. Frenkel, Nano Lett. 19 (2019) 520−529.
Properties of mono- and bimetallic metal nanoparticles (NPs) may depend strongly on their compositional, structural (or geometrical) attributes, and their atomic dynamics, all of which can be efficiently described by a partial radial distribution function (PRDF) of metal atoms. For NPs. Here EXAFS spectroscopy, molecular dynamics simulations, and supervised machine learning (artificial neural-network) method are combined to extract PRDFs directly from experimental data. By applying this method to several systems of Pt and PdAu NPs, we demonstrate the finite size effects on the nearest neighbor distributions, bond dynamics, and alloying motifs in mono- and bimetallic particles and establish the generality of this approach. RMC/EA simulations are used to validate the obtained structure models.
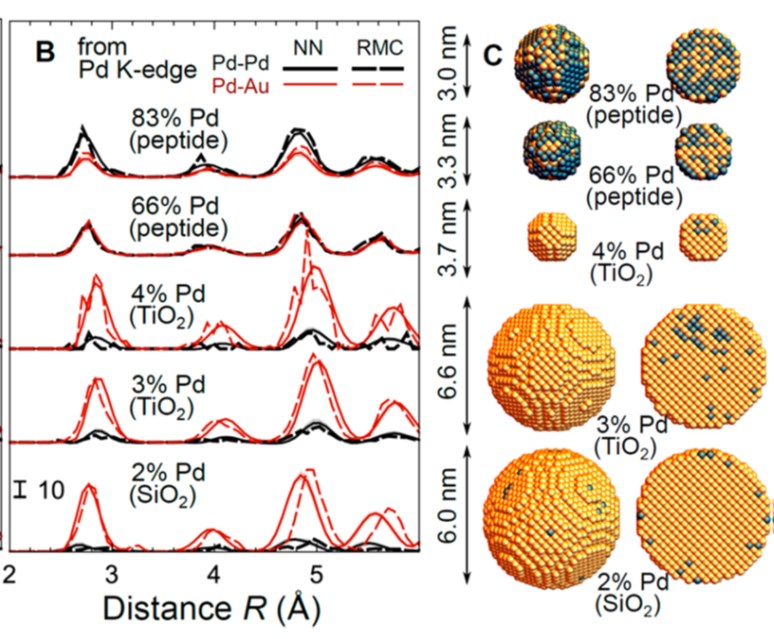
Modeling strain distribution at the atomic level in doped ceria films with extended X-ray absorption fine structure spectroscopy
O. Kraynis, J. Timoshenko, J. Huang, H. Singh, E. Wachtel, A. I. Frenkel, I. Lubomirsky, Inorg. Chem. 58 (2019) 7527−7536.
Ceria doped with trivalent dopants exhibits nonclassical electrostriction, strong anelasticity, and room-temperature (RT) mechanical creep. These phenomena, unexpected for a ceramic material with a large Young’s modulus, have been attributed to the generation of local strain in the vicinity of the host Ce cations due to symmetry-breaking point defects, including oxygen vacancies. However, understanding why strain is generated at the host rather than at the dopant site, as well as predicting these effects as a function of dopant size and concentration, remains a challenge. We have used the RMC/EA to reconcile the EXAFS and X-ray diffraction data in a combined model structure. By extracting the details of the RDF around the host (Ce) and trivalent dopants (Sm or Y), we find that RDF of the first-nearest neighbor (1NN) of host and dopant cations as well as the second-nearest neighbor (2NN) of the dopant are each best modeled with two separate populations corresponding to short and long interatomic distances. This heterogeneity indicates that fluorite symmetry is not preserved locally, especially for the dopant first-and second-NN sites, appearing at surprisingly low doping fractions (5 mol % Sm and 10 mol % Y). Given that Ce rather than dopant sites act as the source of local strain for electrostriction and RT creep, we conclude that the environment around the dopant does not respond to electrical and mechanical excitations, likely because of its similarity to the double fluorite structure which has poor electrostrictive and anelastic properties. The trends we observe in the RDFs around the Ce sites as a function of dopant size and concentration suggest that the response of these sites can be controlled by the extent of doping: Increasing dopant size to increase strain magnitude at the 1NN shell of Ce and decreasing dopant fraction to decrease strain propagation to the 2NN shell of Ce should produce stronger electrostrictive response and RT creep.
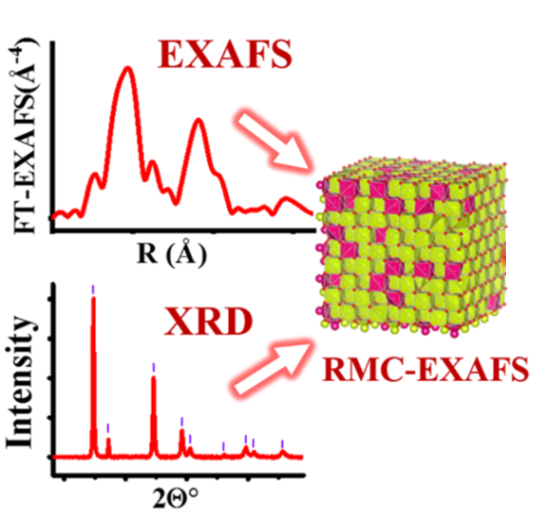
Tuning the structure of Pt nanoparticles through support interactions: An in situ polarized X-ray absorption study coupled with atomistic simulations
M. Ahmadi, J. Timoshenko, F. Behafarid, B. Roldan Cuenya, J. Phys. Chem. C 123 (2019) 10666–10676
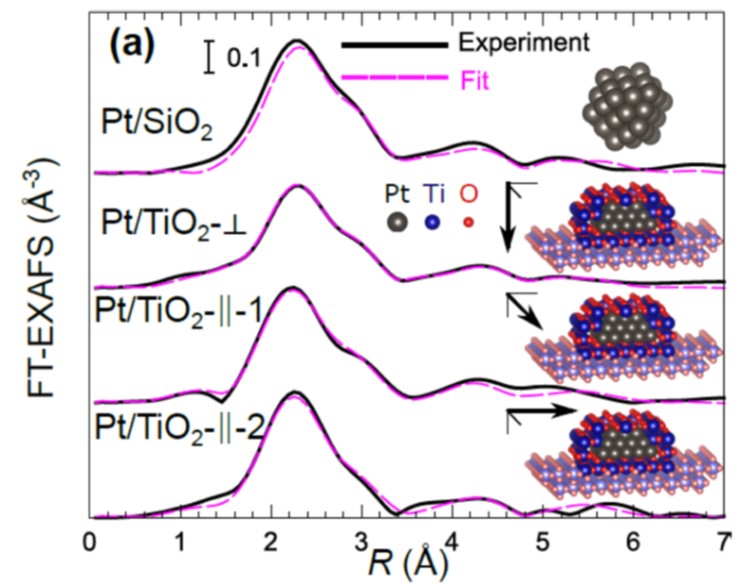
Interactions of nanoparticles (NPs) with their environment may have a pronounced effect on their structure and shape as well as on their functionality in applications such as catalysis. It is therefore crucial to disentangle the particle–adsorbate and particle–support interaction effects on the particle shape, its local structure, atomic dynamics, and its possible anisotropies. In order to gain insight into the support effect, we carried out an XAFS investigation of adsorbate- and ligand-free size-selected Pt NPs deposited on two different supports in ultrahigh vacuum. Polarization-dependent XAFS measurements, neural network-based analysis of XANES data, and RMC/EA simulations of EXAFS were used to resolve the 3D shape of the NPs and details of their local structure. Modeling disorder effects in these NPs using an RMC approach reveals differences in bond-length distributions for NPs on different supports and allows us to analyze their anisotropy, which may be crucial for the interpretation of support-dependent atomic dynamics and can have an impact on the understanding of the catalytic properties of these NPs.
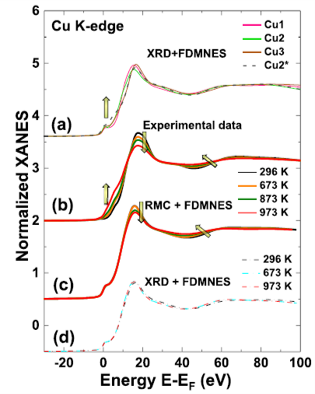
High-temperature X-ray absorption spectroscopy study of thermochromic copper molybdate
I. Jonane, A. Anspoks, G. Aquilanti, A. Kuzmin, Acta Mater. 179 (2019) 26-35.
XAS at the Cu and Mo K-edges was used to study the effect of heating on the local atomic structure and dynamics in copper molybdate (α-CuMoO4) in the temperature range from 296 to 973 K. RMC method was successfully employed to perform accurate simulations of EXAFS spectra at both absorption edges simultaneously. Structure models obtained in RMC-EXAFS simulations were further used to interpret strong temperature-dependence of the Cu Kedge XANES spectra. While the MoO4 tetrahedra behave mostly as the rigid units, a reduction of correlation in atomic motion between copper and axial oxygen atoms occurs upon heating. This dynamic effect seems to be the main responsible for the temperature-induced changes in the O2- to Cu2+ charge transfer processes and, thus, is the origin of the thermochromic properties of α-CuMoO4 upon heating above room temperature.
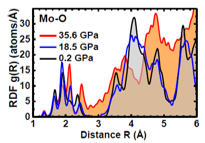
Pressure-induced structural changes in α-MoO3 probed by X-ray absorption spectroscopy
I. Jonane, A. Anspoks, L. Nataf, F. Baudelet, T. Irifune, A. Kuzmin, IOP Conf. Ser.: Mater. Sci. Eng. 503 (2019) 012018.
Energy-dispersive XAS at the Mo K-edge was used to study pressure-induced (up to 36 GPa) changes in the local atomic structure of 2D layered oxide α-MoO3. A linear combination analysis based on the low and high-pressure XANES spectra shows clear evidence of two high-pressure phases, existing at 18-25 GPa and above 32 GPa. The first transition is due to gradual decrease of the interlayer gap, whereas the second one - to its collapse and oxide structure reconstruction. The local atomic structure around molybdenum atoms at 0.2, 18.5 and 35.6 GPa was determined from the EXAFS using RMC calculations.
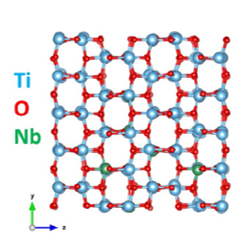
Influence of Nb-doping on the local structure and thermoelectric properties of transparent TiO2:Nb thin films
J.M. Ribeiro, F.C. Correia, A. Kuzmin, I. Jonane, M. Kong, A.R. Goni, J.S. Reparaz, A. Kalinko, E. Welter, C.J. Tavares, J. Alloys Compd. 838 (2020) 155561.
Transparent n-type niobium-doped titanium dioxide thin films with pronounced thermoelectric properties were produced from a composite Ti:Nb target by reactive magnetron sputtering. The local structure of the thin films was investigated in detail by XAS at the Ti and Nb K-edges. A set of radial distribution functions were extracted from the simultaneous analysis of EXAFS data at two absorption edges using the RMC method. It was found that Nb dopant atoms modify the local environment of the films, but their average structure remains close to that of the anatase phase.
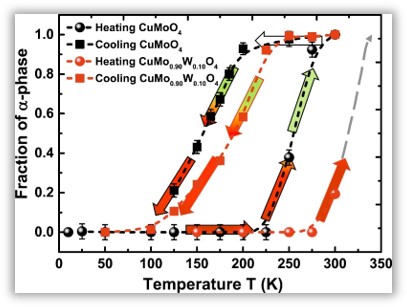
Low temperature X-ray absorption spectroscopy study of CuMoO4 and CuMo0.90W0.10O4 using reverse Monte-Carlo method
I. Jonane, A. Cintins, A. Kalinko, R. Chernikov, A. Kuzmin, Rad. Phys. Chem. 175 (2020) 108411.
Reversible thermochromic phase transition between α- and γ-phases was studied in CuMoO4 and CuMo0.90W0.10O4 using XAS in the temperature range of 10–300 K. RMC/EA approach at several absorption edges simultaneously was applied to extract structural information encoded in the experimental EXAFS spectra. The obtained results show that an addition of 10 mol% of tungsten to CuMoO4 induces local distortions in the structure and stabilizes the γ-phase, leading to an increase of the phase transition temperature by ~50–100 K.
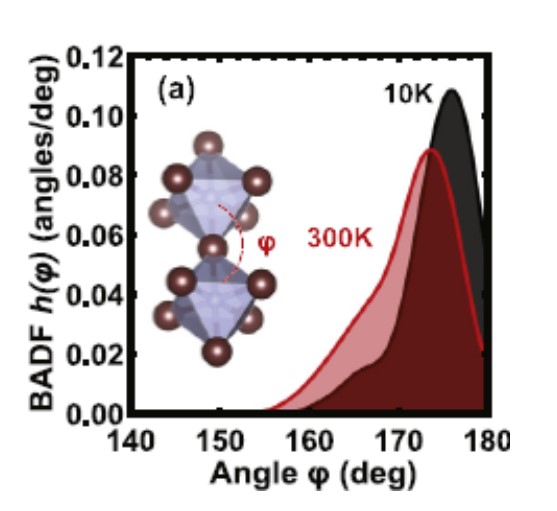
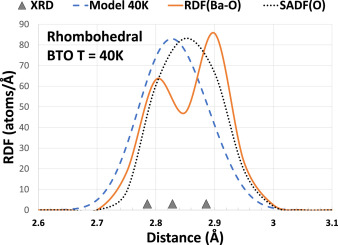
Local structure of A-atom in ABO3 perovskites studies by RMC-EXAFS
A. Anspoks, C. Marini, T. Miyanaga, B. Joseph, A. Kuzmin, J. Purans, J. Timoshenko, A. Bussmann-Holder, Rad. Phys. Chem. 175 (2020) 108072.
The ferroelectric distortions in perovskites were a subject of numerous investigations for a long time. However, some controversial results still exist, coming from the analysis of diffraction (X-ray, neutron or electron) data and XAS. In this study, our goal was to revisit these classical materials using recently developed methods without imposing any predefined structural model. Local environment around A-type atom in ABO3 perovskites (SrTiO3, BaTiO3, EuTiO3) was studied by XAS in a wide range of temperatures (20–400 K). Using RMC/EA, the 3D structure was extracted from the EXAFS and interpreted in terms of the radial distribution functions. Our findings show that both diffraction and XAS data are consistent, but reflect the structure of the material from different points of view. In particular, when strong correlations in the motion of certain atoms are present, the information obtained by XAS might lead to a different from expected shape of the RDF. At the same time, the average positions of all atoms are in good agreement with those given by diffraction.
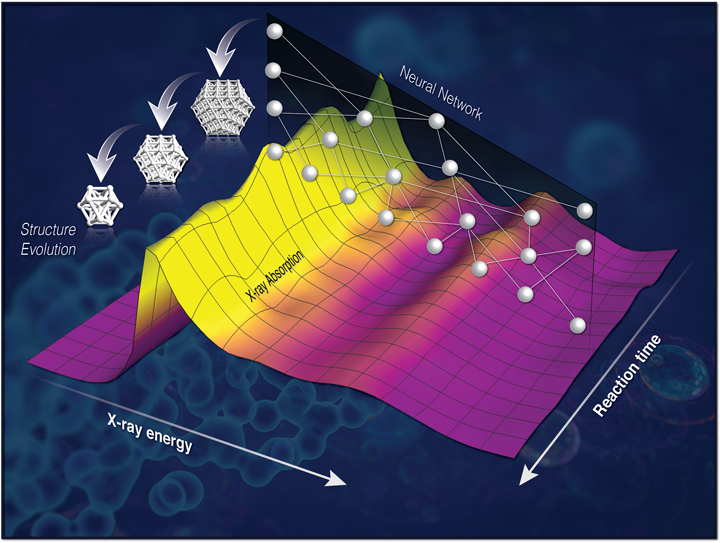
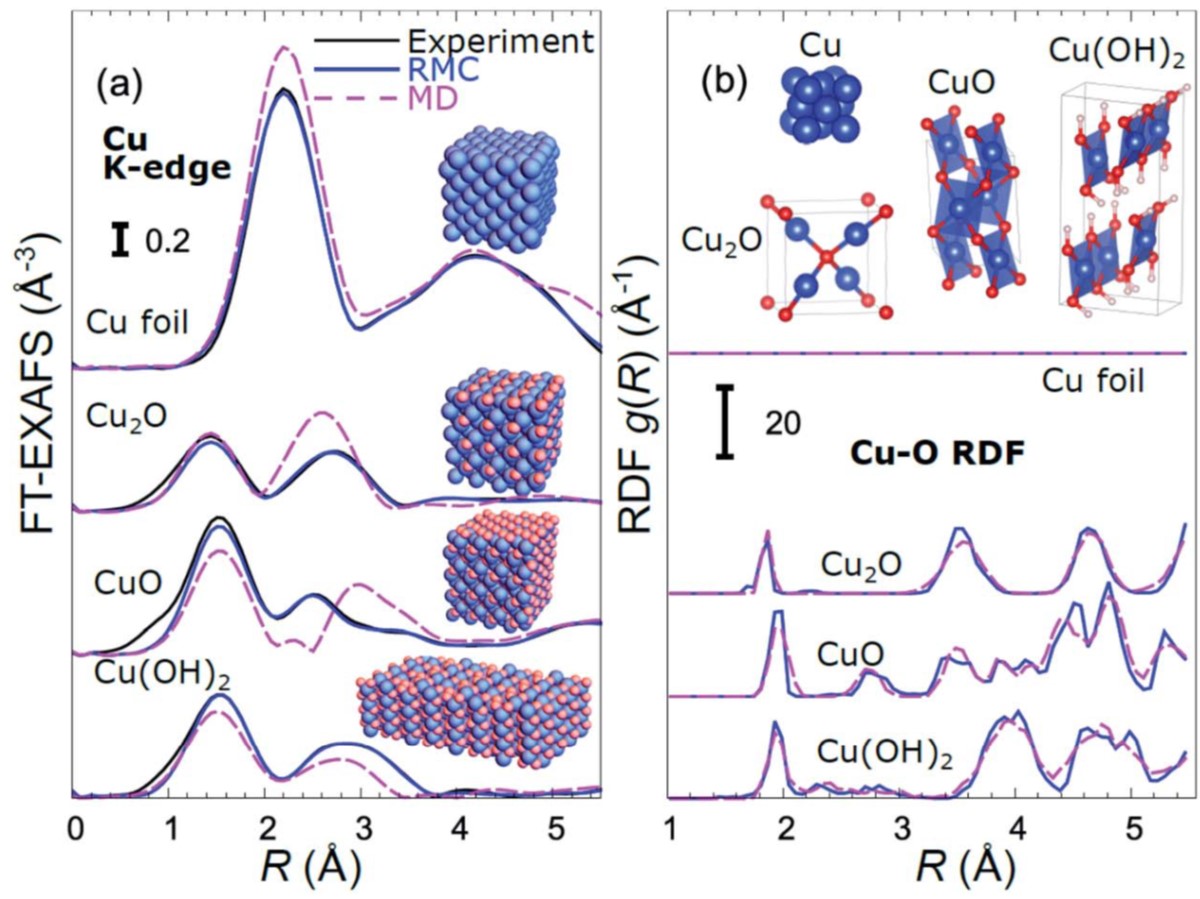
Linking the evolution of catalytic properties and structural changes in copper–zinc nanocatalysts using operando EXAFS and neural-networks
J. Timoshenko, H.S. Jeon, I. Sinev, F. T. Haase, A. Herzog, B. Roldan Cuenya, Chem. Sci. 11 (2020) 3727.
The intrinsic heterogeneity and enhanced disorder characteristic of catalytic materials experiencing structural transformations, as well as the low signal-to-noise ratio that is common for in situ EXAFS spectra hinder the application of conventional EXAFS analysis approaches to probe the catalyst structure under reaction conditions. Here we carry out RMC and molecular dynamics simulations for a large set of Cu- and Zn-based oxides and metals, to establish the relationship between EXAFS features and underlying structure. We then use this information to train an artificial neural network, wich then is used to track time-dependent changes in EXAFS spectra acquired from copper–zinc nanoparticles during the electrochemical reduction of CO2, and to reveal the details of the composition-dependent structural evolution and brass alloy formation, and their correlation with the catalytic selectivity of these materials.
Inner relaxations in equiatomic single-phase high-entropy cantor alloy
A. Smekhova, A. Kuzmin, K. Siemensmeyer, R. Abrudan, U. Reinholz, A. Guilherme Buzanich, M. Schneider, G. Laplanche, K. V. Yusenko, J. Alloy Compd. 920 (2022) 165999.
The superior properties of high-entropy multi-functional materials are strongly connected with their atomic heterogeneity through many different local atomic interactions. The detailed element-specific studies on a local scale can provide insight into the primary arrangements of atoms in multicomponent systems and benefit to unravel the role of individual components in certain macroscopic properties of complex compounds. Herein, multi-edge X-ray absorption spectroscopy combined with reverse Monte Carlo simulations was used to explore a homogeneity of the local crystallographic ordering and specific structure relaxations of each constituent in the equiatomic single-phase face-centered cubic CrMnFeCoNi high-entropy alloy at room temperature. Within the considered fitting approach, all five elements of the alloy were found to be distributed at the nodes of the fcc lattice without any signatures of the additional phases at the atomic scale and exhibit very close statistically averaged interatomic distances (2.54 – 2.55 Å) with their nearest-neighbors. Enlarged structural displacements were found solely for Cr atoms. The macroscopic magnetic properties probed by conventional magnetometry demonstrate no opening of the hysteresis loops at 5 K and illustrate a complex character of the long-range magnetic order after field-assisted cooling in± 5 T. The observed magnetic behavior is assigned to effects related to structural relaxations of Cr. Besides, the advantages and limitations of the reverse Monte Carlo approach to studies of multicomponent systems like high-entropy alloys are highlighted.
More